
what organelle is affected by pompe, check these out | What part of the cell does Pompe disease affect?
By Jessica Wood
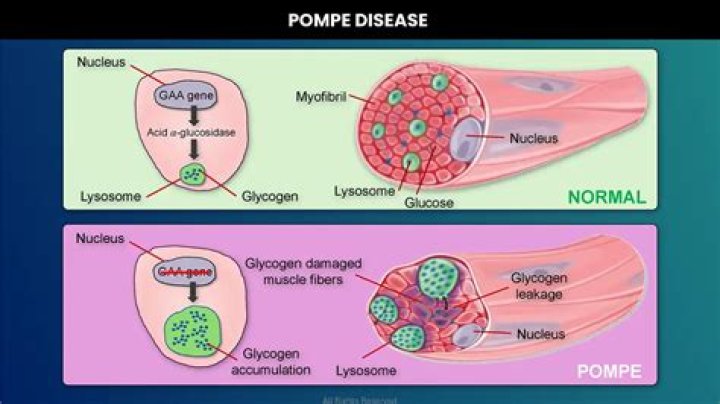
Pompe disease is a lysosomal storage disorder in which acid alpha-glucosidase (GAA) is deficient or absent. Deficiency of this lysosomal enzyme results in progressive expansion of glycogen-filled lysosomes in multiple tissues, with cardiac and skeletal muscle being the most severely affected.
What part of the cell does Pompe disease affect?
In Pompe disease, mutations in the GAA gene reduce or completely eliminate this essential enzyme. Excessive amounts of lysosomal glycogen accumulate everywhere in the body, but the cells of the heart and skeletal muscles are the most seriously affected.
What organelle is affected by glycogen storage disease?
Since mitochondria play a key role in both cellular energy metabolism and apoptosis, it is possible that the function of this organelle is impaired in GSDIa.
How does Pompe disease affect the mitochondria?
We have found multiple mitochondrial defects in mouse and human models of Pompe disease, a life-threatening cardiac and skeletal muscle myopathy: a profound dysregulation of Ca(2+) homeostasis, mitochondrial Ca(2+) overload, an increase in reactive oxygen species, a decrease in mitochondrial membrane potential, an
What role do lysosomes play in Pompe disease?
In Pompe disease, lysosomes do not contain enough of an enzyme called acid alpha-glucosidase (GAA.) This enzyme is necessary to break down glycogen — a complex sugar molecule — into glucose, the simple sugar that the body uses for energy. If glycogen is not broken down, it builds inside cells and causes damage.
What organelle does Pompe disease affect in the cell and how does this disease affect someone’s life?
The defect results in a build-up of glycogen in the lysosome, a saclike storage organelle in the cell that acts as a waste-disposal system, leading to muscle weakness, organ damage including the brain, and possible death.
How does the malfunction of lysosomes affect other organelles?
Dysfunctions of lysosomes can affect the proper activity of other organelles such as peroxisomes and mitochondria, leading to excessive production of reactive oxygen species with pathological features associated with ageing, cancer, chronic inflammation, neurological diseases, male infertility and infections.
What organelle does ALD affect?
Adrenoleukodystrophy (ALD) is a disease linked to the X chromosome. It is a result of fatty acid buildup caused by a defect in the very long chain of fatty acids transporter in peroxisomes, which then causes damage to the myelin sheath of the nerves, resulting in seizures and hyperactivity.
Are lysosomes present in hepatocytes?
In- deed, hepatocyte lysosomes are commonly observed in the vicinity of bile canaliculi, and the fusion between the two can occasionally be recognized morphologically (6, 63).
What organelle is affected by cystic fibrosis?
In most kids with cystic fibrosis, says Balch, the CFTR protein gets stuck inside the cells in a cell organelle known as the endoplasmic reticulum—a convoluted membranous sac within the cell where the synthesis of proteins like CFTR and other vital cell functions take place.
Is Pompe disease a mitochondrial disease?
Pompe disease (PD) is a progressive neuromuscular disorder that is caused by glucosidase acid alpha (GAA) deleterious mutations. Mitochondrial involvement is an important contributor to neuromuscular diseases.
How do people get Pompe?
Pompe disease is inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits a non-working gene from each parent. If an individual receives one working gene and one non-working gene for the disease, the person will be a carrier for the disease.
How do lysosomes interact with other organelles?
Recent research suggests that lysosomes are organelles that store hydrolytic enzymes in an inactive state. The system is activated when a lysosome fuses with another particular organelle to form a ‘hybrid structure’ where the digestive reactions occur under acid (about pH 5.0) conditions.
What role does an enzyme play in Pompe disease?
In Pompe disease the enzyme that is given, acid alpha-glucosidase, breaks down glycogen into glucose, reducing its buildup inside cells and giving cells the energy they need.
What is the biochemistry of Pompe disease?
Discussion. GSD type II (Pompe disease) is an inherited metabolic disorder in which excessive accumulation of glycogen in lysosomes occurs. The mutated (GAA) gene results in a defective enzyme acid GAA, which is responsible for lysosomal degradation of stored glycogen.
Is lysosome an inclusion?
Consequently lysosomes lack the requisite hydrolytic enzymes needed for catabolism of cellular debris, so this debris accumulates within them and forms the characteristic intracellular inclusions (hence the name of the disorder).
What organelle controls movement of material in lungs and fallopian tube?
Microtubules are found in cilia (minute hair‐like organelles involved in movement of mucus out of the lungs and the egg in the fallopian tube), and flagella (similar to cilia but longer and typically found on sperm).
What is Wolman disease?
Wolman disease is a type of lysosomal acid lipase (LAL) deficiency; a rare genetic disorder characterized by complete absence of an enzyme known as lysosomal acid lipase (LIPA or LAL). This enzyme is required to breakdown (metabolize) certain fats (lipids) in the body.
Related Archive
More in general 
harry potter wizarding world japan, latest free online harry potter movies, best HD videos you should watch in 2022 – 2023

harry potter vs voldemort in the deathly hallows, latest free online harry potter movies, best HD videos you should watch in 2022 – 2023

